
- Address
- 305-0044 茨城県つくば市並木1-1 [アクセス]
研究内容
- Keywords
第一原理計算、オーダーN法、プログラム開発
所属学会
日本物理学会, 日本シミュレーション学会
ナノアーキテクトニクス材料研究センター
量子ナノ材料に対する大規模第一原理シミュレーション
第一原理シミュレーション,大規模計算手法,密度汎関数法,機械学習,ナノ複合体
概要
第一原理電子状態計算は物質・材料の原子スケールの構造と物性を定量的に明らかにすることができる強力な研究手法であるが、計算量が膨大で原子を数多く含む複雑大規模系に対する研究を行うことが極めて難しいという問題があった。我々は大規模系を扱うことを可能とする独自の計算手法、プログラムを英国のグループと共同開発し、実材料やデバイスの特性に影響を与える複雑な欠陥構造、表面・界面、そしてナノ構造物質からなる複合体などに対する第一原理計算にもとづいた理論研究を行なっている。開発されたプログラムは様々な物質・材料に適用可能で、多くの研究者が用いることができるように無償公開されている。
新規性・独創性
● 通常の第一原理計算が扱えない大規模系の計算が可能
● 従来手法よりも数十倍から1千倍程度の原子数を扱うことが可能
● 英国のグループ(MANAサテライト)と共同開発したプログラムの一般公開
● 機械学習力場と第一原理計算を同時に用いた複雑大規模系への理論研究
● 複雑な系における局所構造を解析するための独自の機械学習手法
内容
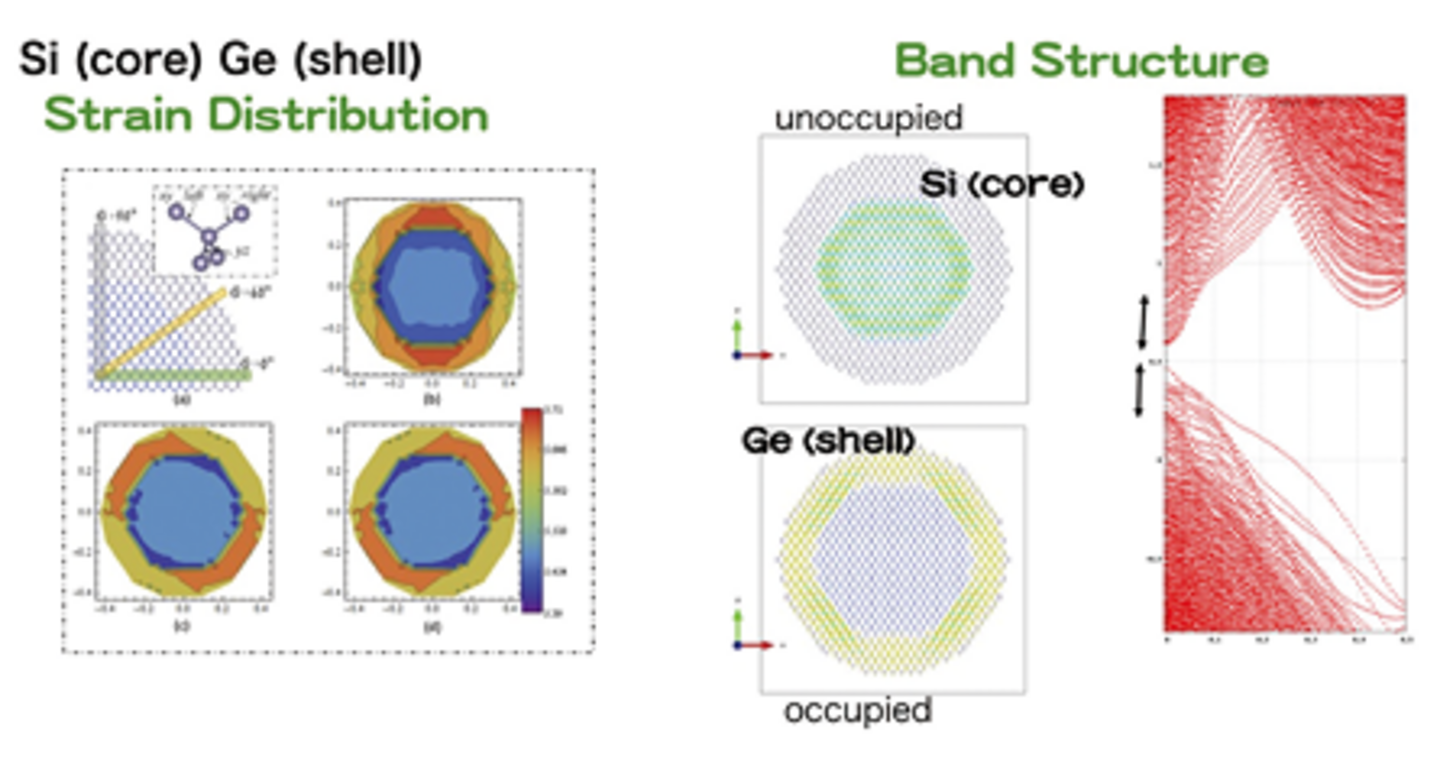
我々は英国の研究グループと共同で大規模第一原理計算プログラムCONQUESTの開発を行ってきた。このプログラムはオーダーN法や独自の局在軌道法を用いることによって、通常の第一原理計算手法では扱えないような巨大系に対しても第一原理計算に基づいた電子状態解析、構造最適化、分子動力学(MD)を行うことが可能となっている。CONQUESTを用いた研究例として、半導体コアシェル型ナノワイヤに対する大規模第一原理計算による理論解析が図に示されている。現実のナノワイヤのサイズを持つ構造モデル(原子配置)に対する構造最適化計算により、コア領域のシリコンとシェル領域のゲルマニウムの格子定数の違いから生じるナノワイヤ内の歪み分布、さらにフェルミ準位付近の電子状態(キャリア)の分布などを定量的に明らかにすることに成功している。プログラムCONQUESTは2020年1月に寛容型オープンソースライセンスであるMITライセンスにより公開された。これにより、国内外の研究者がこのプログラムを無償で用いることが可能となっている。
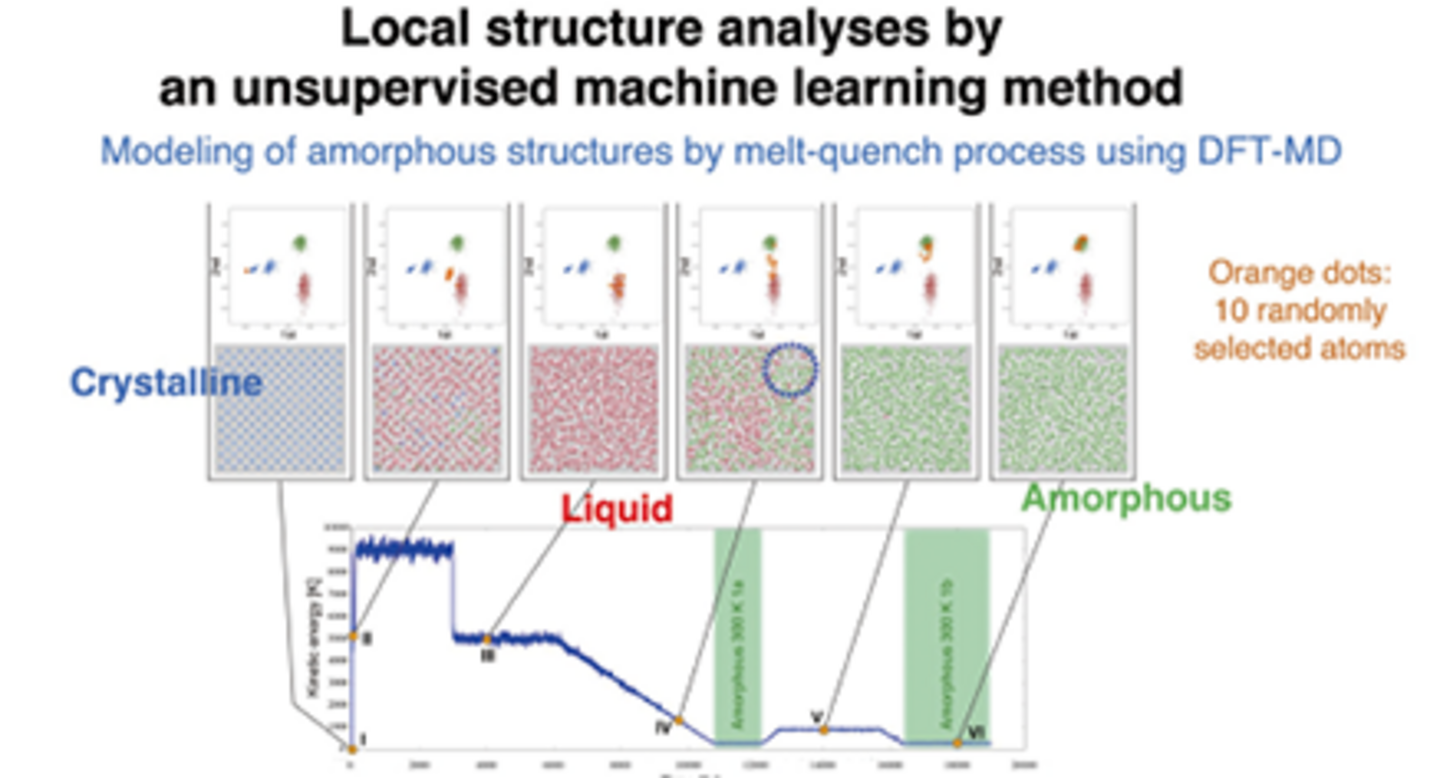
また、我々のグループではCONQUESTによる大規模第一原理MDを表面、欠陥、非晶質構造などに適用している。これら複雑系の分子シミュレーションにおいて現れる様々な構造を理解し、その結果現れる物性との相関を明らかにすることが重要な研究課題となっている。我々は分子シミュレーションで現れる様々な局所構造を教師無しの機械学習にもとづいて解析する新しい手法を提案し、その有効性を示すことに成功した。シリコンのアモルファス構造を作成するためのMelt-Quench過程に対する解析が右図に示されている。現在、様々な物質群にこの手法を適用し、その有効性を調べている。今後、この手法をさらに発展させ、すぐれた機能を持つ物質をデザインしていくことを目指している。
まとめ
我々が開発したプログラムCONQUESTにより、今までは不可能であった大規模複雑系に対しても第一原理計算による研究が可能となった。第一原理計算は様々な物質に適用可能で、我々は今までに半導体ナノ表面構造、コアシェル型ナノワイヤ、金属ナノ微粒子、強誘電体ドメイン構造、シリカガラス、水溶媒中の巨大生体分子等に対して研究を行なってきた。今後、より広範囲の物質群、現象に適用していく予定である。