
- Address
- 305-0044 茨城県つくば市並木1-1 [アクセス]
- Web Site
研究内容
出版物2004年以降のNIMS所属における研究成果や出版物を表示しています。
エネルギー・環境材料研究センター
高スループット第一原理計算とAI手法による全固体電池用の固体電解質材料デザイン
全固体電池,固体電解質,第一原理計算,分子動力学計算,マテリアルズインフォマティクス,機械学習
概要
全固体電池は、非可燃性の固体電解質と高電圧の正極材料または高容量の電極の組み合わせにより、高エネルギー密度と高い安全性を備えているため、次世代のエネルギー貯蔵において最も有望な候補として広く認識されている。特に、全固体電池はの優れた特徴は、固体電解質の高い単一イオン伝導性、優れた熱安定性、簡素化されたパッケージング設計に由来している。本研究では、高スループット第一原理計算とAI手法を用いて、Liイオン電導度の高い固体電解質の新規材料探索を行う。
新規性・独創性
本研究は、高精度の第一原理の分子動力学計算を用いて材料物性(例:Liイオン伝導度)を計算し、AIベースの材料物性最適化手法に基づいて広範な材料探索空間で優れた新しい化合物を系統的に探索すること。そして、イオン伝導度だけでなく、材料の熱力学的安定性も評価し、現実的な候補材料を提案することができる。
内容
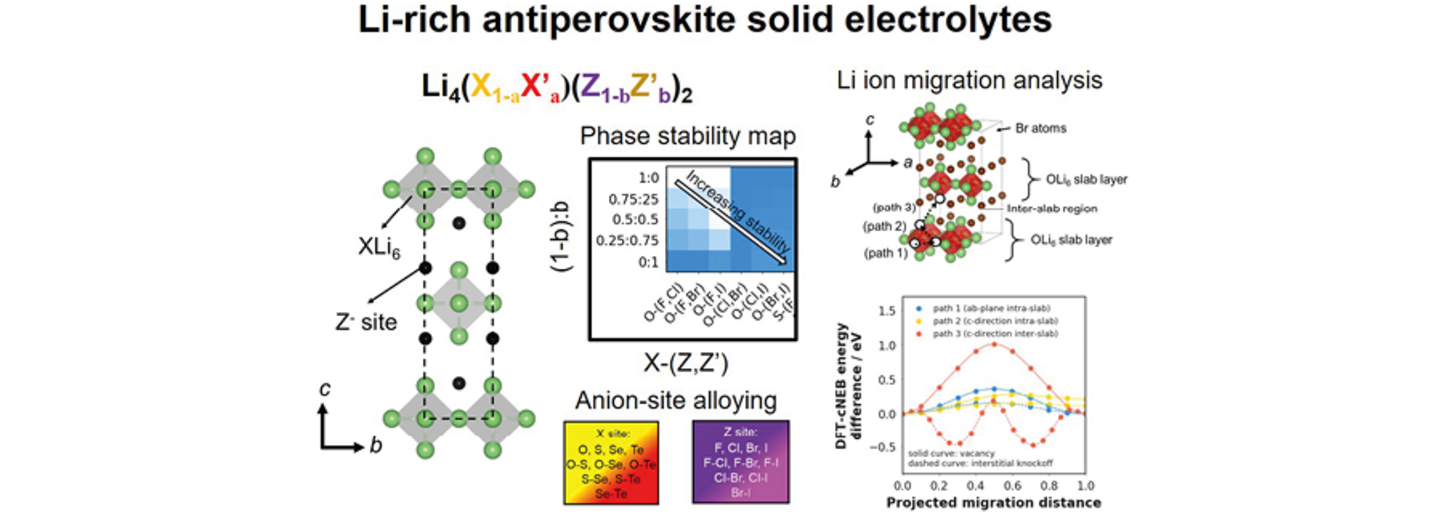
近年、Liリッチの逆ペロブスカイト構造を持つ化合物は、全固体電池用の固体電解質として大きな関心を集めている。逆ペロブスカイト構造を持つ候補固体電解質の報告はまだ比較的少ないですが、このような材料に存在する結晶系や構造の派生物は非常に多様である。本研究では、密度汎関数理論の計算を用いて、一般式Li4(X1–aX'a)(Z1–bZ'b)2(X、X' ∈ {O2–、S2–、Se2–、Te2–};Z、Z' ∈ {F–、Cl–、Br–、I–};0 ≤ a, b ≤ 1)を持つ逆ペロブスカイト構造の化合物500以上の材料スクリーニングを行った。固体電解質としての候補新規化合物の特定、固体電解質設計の有用な記述子の明確化、およびこの系における特徴的なLiイオン拡散メカニズムの決定を目指した。約167の化合物が、分解エネルギーが0.1 eV/原子以下で熱力学的に安定・準安定であると予測された。特に、Li4O(Cl1–bBrb)2系、酸素/ヨウ素を含む組成、および酸素/硫黄を含む組成に属する少なくとも20の新規化合物を強調した。Liリッチの逆ペロブスカイト化合物の熱力学的安定性と電子バンドギャップエネルギーの良い記述子として、Goldschmidt許容因子の改良された形式が見つかった。一方、イオンの経路の空隙空間マップから抽出した幾何学的特徴は、イオン輸送特性の有用な記述子として特定された。第一原理分子動力学計算に基づいて、代表的な2つの化合物であるI4/mmm Li4OBr2とCmcm Li16O3SI8のバルクLi+イオン活性化エネルギーはそれぞれ0.29および0.46 eVと推定された。
まとめ
有望なインシリコ候補固体電解質材料の実際の合成は現在進行中です。また、大規模な材料スクリーニングのために開発された計算手法は、固体電解質の探索だけでなく、他のバッテリー材料の探索にも使用される予定である。生成されたデータセットは、将来のデータ駆動型材料設計の取り組みにおいても非常に有用と期待されている。