研究内容
- Keywords
[4/26更新]First-Principles,Band calculation,Search for unknown materials(Materials Design), BN,AlN,SiC,Polytype,Layer structure,TiN/MgO(Surface and Interface),Electronic Structure,Electronic Band Structure,Wide Band Gap,Car-Parrinello method,Pseudopotential,Plane wave basis sets,DFT,LDA,Pseudopotential Database,NCPS2K,

[QRコードテスト](左横にあるのと違う古いもの[アクセス可能])

[筆者の今日の予定]:特になし(4/26、2024)。
[筆者の近辺の予定]:特になし(4/26、2024)。
[4/30]会合(オンライン、午後、予定)
[5/26]NIMS一般公開2024(並木/千現/桜)
[7/9]会合(東京、午後、予定)
[7/30]会合(並木、午後、予定)
[6/15~19、2025]ICT/ACT2025(仙台)
[BEYOND THE 2C PROJECT][バンド計算関連情報][暦][地図](国内、国際会議、記事
等、[↑外部ページ↑])
[New papers]
[1]Cédric Bourgès, Wenhao Zhang, Krushna Kumari Raut, Yuka Owada, Naoyuki Kawamoto, Masanori Mitome, Kazuaki Kobayashi, Jean-François Halet, David Berthebaud, and Takao Mori, "Investigation of Mn Single and Co-Doping in Thermoelectric CoSb3-Skutterudite: A Way Toward a Beneficial Composite Effect", ACS Appl. Energy Mater. 2023, 6, 18, 9646–9656.
[2]Kazuaki Kobayashi, Hirokazu Takaki, Masato Shimono, Hiroyuki Ishii, Nobuhiko Kobayashi, Kenji Hirose, and Takao Mori, "Electronic and magnetic properties of CoSb3, Cr-doped CoSb3, and related compound thin films", Jpn. J. Appl. Phys. 62[SC] (2023) SC1046 (Special issue).
[3]Kazuaki Kobayashi, Hirokazu Takaki, Masato Shimono, Hiroyuki Ishii, Nobuhiko Kobayashi, Kenji Hirose, Naohito Tsujii and Takao Mori, "First-principles study of Fe2VAl and Fe2VAl/Si thin films and their magnetic properties", Jpn. J. Appl. Phys. 61[SL] (2022) SL1013 (Special issue).
[4]Takuro Tokunaga, Masao Arai, Kazuaki Kobayashi, Wataru Hayami, Shigeru Suehara, Takuma Shiga, Keunhan Park, and Mathieu Francoeur, "First-principles calculations of phonon transport across a vacuum gap", Phys. Rev. B 105, 045410 (2022).
[5]Kazuaki Kobayashi, Hirokazu Takaki, Masato Shimono, Nobuhiko Kobayashi, and Kenji Hirose, "Electronic and Lattice Properties of Nanostructured TiN/MgO and ScN/MgO Superlattices", Jpn. J. Appl. Phys. 60[SE] (2021) SE1006 (Special issue).
[6]Jean-Baptiste Vaney, Sima Aminorroaya Yamini, Hirokazu Takaki, Kazuaki Kobayashi, Nobuhiko Kobayashi, Takao Mori, "Magnetism-mediated thermoelectric performance of the Cr-doped bismuth telluride tetradymite", Mater. Today Phys. 8 (2019) 100090.
[7]Hirokazu Takaki, Kazuaki Kobayashi, Masato Shimono, Nobuhiko Kobayashi, Kenji Hirose, Naohito Tsujii, and Takao Mori, "Seebeck coefficients in CuFeS2 thin films by first-principles calculations", Jpn. J. Appl. Phys. 58[SI] (2019) SIIB01 (Special issue).
[8]Kazuaki Kobayashi, Hirokazu Takaki, Masato Shimono, Nobuhiko Kobayashi, and Kenji Hirose, "Electronic Band Structure of TiN/MgO-4×4 and 5×5 Nanostructures", Jpn. J. Appl. Phys. 58[SB] (2019) SBBH06 (Special issue).
[9]Hirokazu Takaki, Kazuaki Kobayashi, Masato Shimono, Nobuhiko Kobayashi, Kenji Hirose, Naohito Tsujii, and Takao Mori, "Thermoelectric properties of a magnetic semiconductor CuFeS2", Mater. Today Phys. 3 (2017) 85-92.
[10]Hirokazu Takaki, Kazuaki Kobayashi, Masato Shimono, Nobuhiko Kobayashi, and Kenji Hirose, "Enhancement of thermoelectric properties in surface nanostructures", J. Electron. Mater. 46[10], 5593 - 5598 (2017).
[11]Kazuaki Kobayashi, Atta Ullah, and Takao Mori, "Electronic structures of Si- and Te-doped CoSb3 compounds under high pressures", Jpn. J. Appl. Phys. 56[5S3] (2017) 05FB07 (Special issue).
[12]Kazuaki Kobayashi, Hirokazu Takaki, Masato Shimono, Nobuhiko Kobayashi, and Kenji Hirose, "Electronic band structure of TiN/MgO nanostructures", Jpn. J. Appl. Phys. 56[4S] (2017) 04CK06 (Special issue).
[13]Hirokazu Takaki, Kazuaki Kobayashi, Masato Shimono, Nobuhiko Kobayashi, Kenji Hirose, Naohito Tsujii, and Takao Mori, "First-principles calculations of Seebeck coefficients in a magnetic semiconductor CuFeS2", Appl. Phys. Lett. 110[7] (2017) 072107.
[14]A. U. Khan, K. Kobayashi, D. Tang, Y. Yamauchi, K. Hasegawa, M. Mitome, Y. Xue, B. Jiang, K. Tsuchiya, D. Golberg, Y. Bando, and T. Mori, "Nano-micro-porous skutterudites with 100% enhancement in ZT for high performance thermoelectricity", Nano Energy, 31, 152 - 159 (2017).
[15]H. Takaki, K. Kobayashi, M. Shimono, N. Kobayashi and K. Hirose, "First-principles calculations of thermoelectric properties of TiN/MgO superlattices - the route for enhancement of thermoelectric effects in artificial nanostructures", J. Appl. Phys. 119 (2016) 014302.
[16]K. Kobayashi, H. Takaki, N. Kobayashi, and K. Hirose, "Electronic Band Structure of Various TiN/MgO Superlattices", JPS Conf. Proc. 5, 011013(2015) (CSW2014)
[17]K. Kobayashi, N. Kobayashi, and K. Hirose, "First-Principles Study of TiN/MgO Interfaces", e-J. Surf. Sci. Nanotech., 12 (2014) 230 - 237 (ACSIN-12 & ICSPM21)
[18]K. Kobayashi and S. Komatsu, "First-Principles Study of AlBN and Related Polytypes", Trans. MRS-J, Vol. 38[3], 485-492 (2013).
[19]K. Kobayashi and S. Komatsu, "First-Principles Study of Various BN, SiC, and AlN polytypes", Trans. MRS-J, Vol. 37, 583-588 (2012)[IUMRS-ICEM2012].
[20]K. Kobayashi and S. Komatsu, "First-Principles Study of 8H-, 10H-, 12H-, and 18H-SiC Polytypes", Journal of the Physical Society of Japan, Vol. 81, No. 2 (2012) 024714.
[My Portal](ReaD & Researchmap)
[公開版]12月24日(2010)に行われたイブニング
セミナーの発表原稿(PDF形式、約3MB、無保証)
第一原理とは、ここでは「何ら実験結果に依らない。」という意味である(研究分野や、研究者個々で解釈が”微妙”に異なる場合あり)。 [関連語]第一原理計算、ab initio
バンド計算とは、バンド理論に基づいて、固体の電子状態(電子構造)を数値的に解く一方法。現在は、第一原理に基づいたバンド計算手法が主流(第一原理でないバンド計算手法も存在する)。 現在では、固体以外(例:表面、界面など)も計算対象にすることが可能。
カー・パリネロ法(Car-Parrinello method)とは、電子状態計算(バンド計算)と分子動力学計算を融合した手法(日本語では、”第一原理分子動力学計算”と呼ばれることが多い)。従来のバンド計算では行列要素(HΨ=EΨのH 部分)を対角化(固有値問題)で解いていたが、カー・パリネロ法は対角化を用いずに数値的な逐次計算で固有ベクトル(電子の波動関数に相当)と固有値を求める。同時に構造の最適化(単位胞及び単位胞内の原子の安定構造を求めること)を行うことも可能にした。
バンド構造とは、主に固体の電子状態を表現する手段の一つ。ブリュアン・ゾーン内のk点とそれに対応する電子のエネルギー固有値E(k)との関係を図として描いたもの。
バンド分散、E-k曲線、E-k分散などとも言う。勿論、バンド計算出来れば、固体以外のバンド構造も描くことが可能。[関連語]状態密度、バンドギャップ、バンド理論
[参考ページ]バンド構造図集、周期表
ポリタイプ(polytype)とは、一次元の多形構造のこと。ポリタイプとしては、SiC(シリコンカーバイド、炭化ケイ素)が有名。ポリタイプに関するより詳しい情報は、BNのページ参照。
擬ポテンシャルとは、原子において価電子部分のみを考慮したポテンシャル。これは固体の性質(物性)の多くが、実質上価電子のみからの寄与で説明可能だからである。勿論、内殻電子からの寄与が重要となる場合もある(そのよ
うな場合には、通常の擬ポテンシャルでは扱うことが難しくなる)。ノルム保存型や、超ソフト型などいくつかの種類がある。ノルム保存型や超ソフト型は非経験(=第一原理)的に作成される。[関連語]全電子計算、平面波
並列計算とは、一つのプログラムを複数のCPU(または計算機)で並列に実行させること。並列計算させるためには、対象となるプログラムを並列計算用に作成するか、そ
うなるように改良する必要がある。並列化する手法として、OpenMP(共有メモリ上で並列実行)、MPI(分散メモリ上で並列実行可能)などがある。[関連語]スカラー、ベクトル化、クラスター、GPGPU、スーパーコンピュータ、対義語:逐次実行
2024年度(本年度)計画:スクッテルダイト系、その他の薄膜構造の計算など進行中。
2023年度(前年度)計画:VN/MgO系、ホイスラー系、スクッテルダイト系、その他の薄膜構造の計算など進行中。
2022年度(前々年度)計画:引き続き擬ポテンシャルデータベースの拡充(希土類等への対応など)作業を進める予定。その他はいろいろ計画(VN/MgO系、ホイスラー系、その他の薄膜構造の計算など)進行中。
2021年度(ほんの少し前)計画:引き続き擬ポテンシャルデータベースの拡充(希土類等への対応など)作業を進める予定。その他はいろいろ計画(VN/MgO系、その他の薄膜構造の計算など)が進行した。
2020年度(ほんの少し昔)計画:引き続き擬ポテンシャルデータベースの拡充(希土類等への対応など)作業を進めた。2020年度は、データの可視化に進展があった。その他はいろいろ計画(TiN/MgO、ScN/MgO、VN/MgO系の計算など)を進行させた。新年度早々から、新型コロナウィルスの影響で研究進捗に大きな支障があった。
2019年度(ちょっと昔)計画:引き続き擬ポテンシャルデータベースの拡充(希土類等への対応など)作業を進める予定だったが、現状維持で精一杯な状況が続いた。その他はいろいろ計画(TiN/MgO、ScN/MgO系の計算など)が進行した。
2018年度(少し昔)計画:引き続き擬ポテンシャルデータベースの拡充(希土類等への対応など)作業を進めた。その他はいろいろ計画(TiN/MgO-4x4, 5x5超格子系の計算など)が進行した。
2017年度(少し昔)計画:引き続き擬ポテンシャルデータベースの拡充(希土類等への対応など)作業を進めた。その他はいろいろ計画が進行した。
2016年度(昔)計画:引き続き擬ポテンシャルデータベースの拡充(希土類等への対応など)作業を進める予定だったが進捗は芳しくない。その他はいろいろ計画(TiN/MgOナノ構造計算、CoSb3関連物質の計算)が進行した。
2015年度(もっと昔)計画:引き続き擬ポテンシャ ルデータベースの拡充(希土類等への対応など)作業を進める。その他はいろいろ進行した。
2014年度(大分昔)計画:引き続き擬ポテンシャルデータベースの拡充(希土類等への対応)作業を進める。三元系ポリタイプや更に大きなポリタイプ構造の計算を行う。
2013年度(大昔)計画:引き続き擬ポテンシャルデータベースの拡充(希土類等への対応)作業を進める。更に大きなポリタイプ構造の計算を行う。
2012年度(大昔)計画:擬ポテンシャルデータベースの拡充(希土類等への対応)作業を進めた。20H-SiC,48H-BN,30H-AlNなどの大きなポリタイプの計算を行った。
2011年度(大昔)はBN,SiC,AlNポリタイプ構造において特にSiCポリタイプを中心にして、その電子状態(電子構造)と安定構造を第一原理分子動力学手法を使って求めた。計算の結果、10H-SiC(ABCACBCACB)が計算したSiCポリタイプの中で最も安定であることが分かった。詳細は、本ページでも紹介しているBNのウェブページを参照して欲しい。更にノルム保存型擬ポテンシャルデータベース NCPS2Kの配布を行っている。
2H~6Hまでのポリタイプをいろいろな記法で表現
| - | ABC記法 | Hägg記法 | h-c記法 | Zhdanov記法 | H(%) |
|---|---|---|---|---|---|
| 2H | AB | +- | hh | 11 | 100 |
| 3H | ABC | +++ | ccc | 3(∞) | 0 |
| 4H | ABCB | ++-- | hchc | 22 | 50 |
| 5H | ABCBC | ++++- | hhccc | 41 | 40 |
| 6H | ABCACB | +++--- | hcchcc | 33 | 33.3 |
| 6H | ABCBCB | ++-+-- | hchhhc | 2112 | 66.7 |
尚、筆者のその他の研究活動や
第一原理計算(バンド計
算)全般の情報に関しては、筆者公式
ページ(とそこか
ら辿れるページ、サイト)を参照して欲しい。
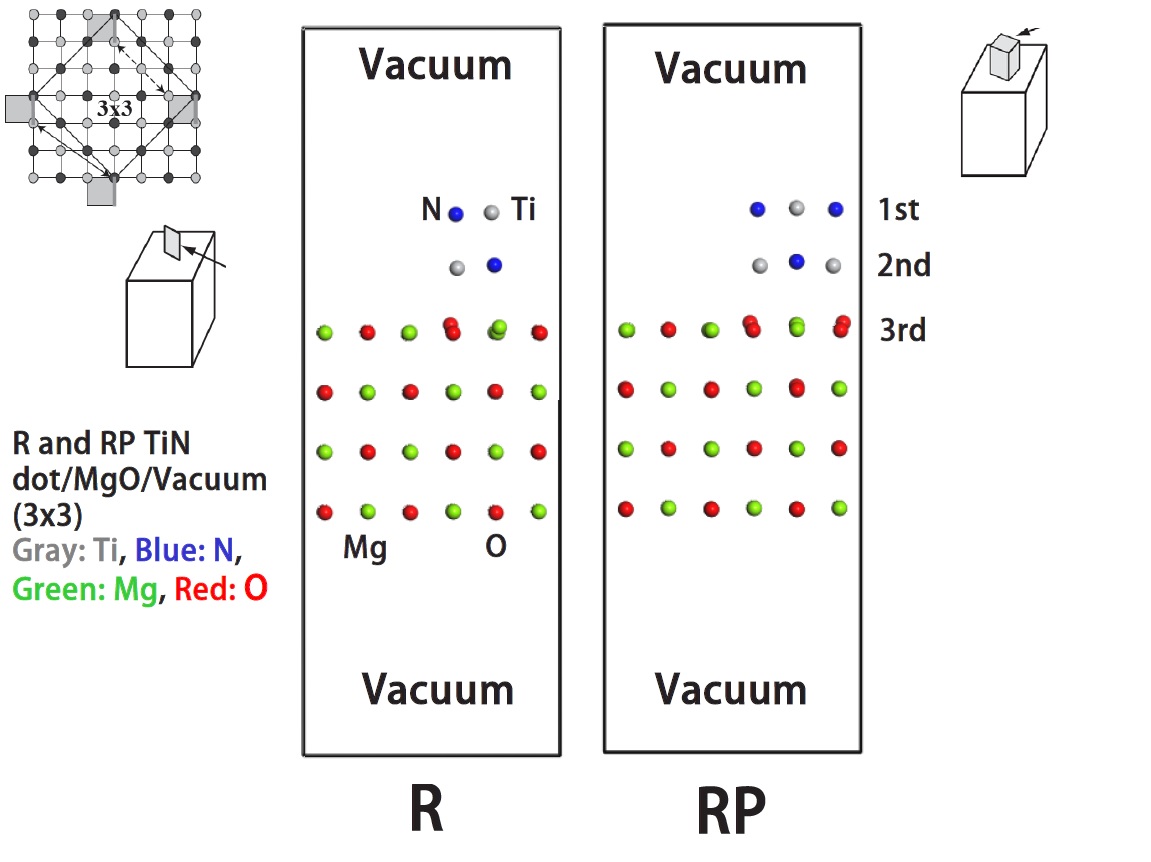
Side views of relaxed structures for rectangular and rectangular parallelepiped TiN dot/MgO-3x3 superlattices.
所属学会
[4/26]日本物理学会,応用物理学会,日本計算工学会,日本コンピュータ化学会