HOME > Profile > NAKATA, Ayako

- Address
- 305-0044 1-1 Namiki Tsukuba Ibaraki JAPAN [Access]
Research
- Keywords
Quantum chemistry, First-principles calculation, Large-scale DFT simulation
Society memberships
理論化学会, 日本化学会, 日本物理学会
Research Center for Materials Nanoarchitectonics (MANA)
大規模第一原理DFT計算の開発と応用
密度汎関数(DFT)法,電子状態計算,金属ナノ粒子触媒
Overview
第一原理DFT計算を用いた構造・電子状態のシミュレーションは、有機・半導体・金属など様々な材料の性質の解析・予測に用いられ、新規材料の開発においても非常に有力なツールである。そのためには実在の環境に近いナノスケールでのシミュレーションが求められる。本研究では、当グループで開発している大規模DFT計算プログラムCONQUESTをもとに、新たな高効率・高精度な計算手法の開発・導入を行うことによって、DFT計算をナノサイズの材料に適用するための開発を行う。またその手法を用いて、ナノスケール実材料への応用を目指す。
Novelty and originality
● 高精度・高効率な大規模第一原理計算手法の開発
● 大規模DFT計算の解析手法の開発
● 複雑な構造をもつ材料(ナノスケール表面、界面、不規則系)への応用
● 金属ナノ粒子触媒などナノスケール実材料への応用
Details
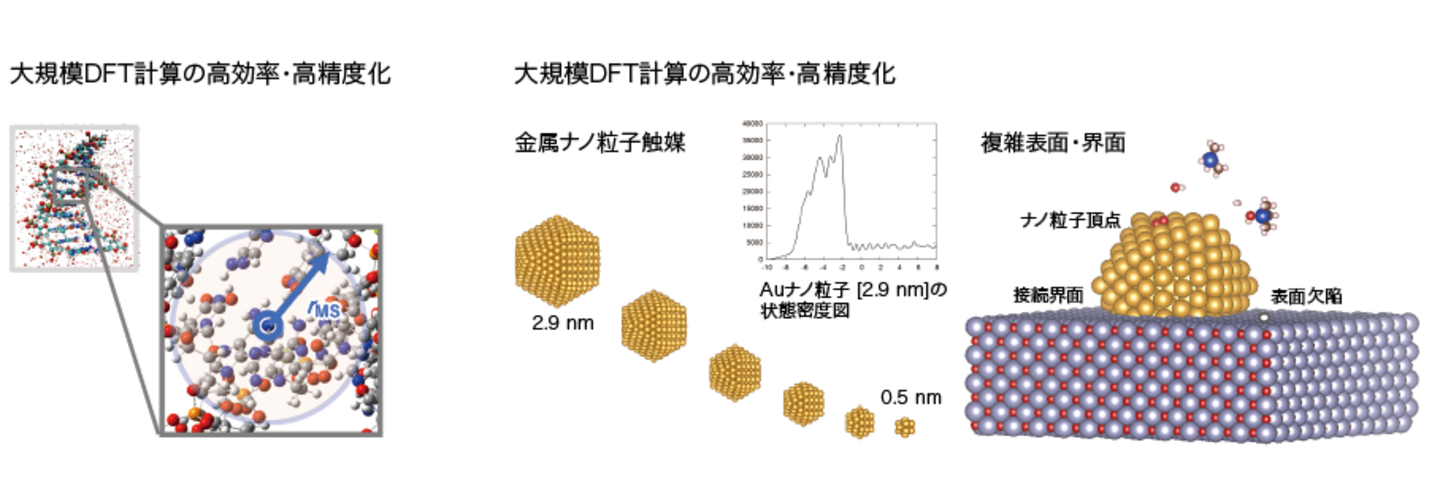
新たな基底関数の縮約手法(マルチサイト法)の導入により、第一原理計算の低コスト化・精度の向上に成功している。この方法を用いることで、金属、半導体、絶縁体を問わず幅広い材料に関する大規模第一原理計算が可能となっている。
この方法を用いて、例えば、実際に用いられている数nmサイズの金属ナノ粒子触媒に関して、構造や電子状態、反応性の詳細な解析に取り組んでいる。大規模計算では計算結果も煩雑となるため、統計解析や機械学習との組み合わせによって大規模系の中から特徴的な箇所を効率よく探索することで、ナノスケール材料の高精度、高効率な理論解析を目指している。
Summary
● 大規模DFT計算の低コスト化・高精度化のための手法を開発
● 大規模DFT計算によって得られる大規模系の構造や電子状態を効率よく解析するための手法を提案
● ナノスケール材料における複雑表面・界面の構造・電子状態解析への応用